KIELTYKA GLADKOWSKI KG LEGAL TOOK PART IN LIFE SCIENCE CLUSTER WEBINAR ON CERTIFICATION OF MEDICAL DEVICES
Publication date: March 31, 2025

The meeting was devoted to the classification of medical devices. The main topic was the technical and legal classification and certification of medical products and diagnostic devices before they are introduced to the market. This process is very time-consuming and even very expensive.
It is worth using the help of a company that provides assistance in this procedure during this process. It is important that this company is a notified body, a conformity assessment body designated in accordance with Regulation 2017/745. Then you can be sure that a given product intended to take care of human health will be properly put into clinical trials and will be safe for people.
What is a medical device?
A medical device is a tool, apparatus, device, software, implant, reagent, material or other article intended by the manufacturer for use in humans for medical purposes. It may be used alone or in combination for one or more of the following specific medical purposes:
- Diagnosing, preventing, monitoring, predicting, prognosing, treating or mitigating disease.
- Diagnosing, monitoring, treating, alleviating, or compensating for an injury or disability.
- Studying, replacing, or modifying an anatomical structure or a physiological or disease process or condition.
- Providing information through in vitro examination of samples collected from the human body, including those from organ, blood and tissue donors.
A medical device does not achieve its principal intended action by pharmacological, immunological or metabolic means in or on the human body, but its function may be assisted by such means.
The following products are also considered medical devices:
- Products for the purpose of conception control or assistance.
- Products specifically designed for cleaning, disinfecting or sterilizing medical devices.
The process of preparing a medical device for assessment of compliance with the previously mentioned regulation.
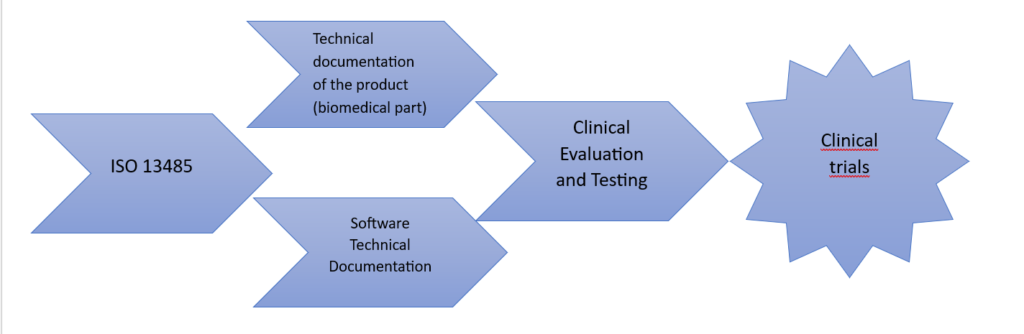
ISO 13485 – Quality Management System for Medical Devices
ISO 13485, entitled “Medical devices – Quality management systems – Requirements for regulatory purposes”, specifies requirements for a quality management system that can be applied by organizations engaged in the design, development, production, installation and servicing of medical devices and the provision of related services.
ISO 13485 can be implemented independently, as it is its first stand-alone version, but due to its links with ISO 9001, it is also possible to use it in combination with this standard. As a result, organizations can obtain certificates of conformity with ISO 13485 or with ISO 9001 and ISO 13485 at the same time.
The international standard ISO 13485 specifies the requirements for a quality management system for companies that must demonstrate their ability to supply medical devices and related services that fully meet legal requirements and customer expectations.
The standard implementation process is similar to ISO 9001, but with greater emphasis on industry regulations and risk analysis related to potential threats to users of medical devices.
The basic idea of ISO 13485 is to harmonize the legal requirements for medical devices with the quality management system. Therefore, this standard includes additional requirements specific to this industry, while omitting some elements contained in ISO 9001 that do not refer to legal regulations. For this reason, organizations with a quality management system compliant with ISO 13485 cannot automatically recognize it as compliant with ISO 9001 unless they also meet all the requirements specified in the latter standard.
Manufacturers of products other than custom-made products are required to maintain technical documentation, the requirements of which are as follows:
1. Product description and specification
- Name and purpose – trade name, description, users and applications.
- Identification – Basic UDI-DI code or other number ensuring traceability.
- Intended population – diseases, indications, contraindications, warnings.
- Principles of operation – method of operation, scientific justification.
- Classification and risk – justification for the assigned product class.
- New Features – Any innovation in design or operation.
- Equipment and configurations – list of variants and additional elements.
- Materials and composition – description of raw materials, technical features and graphical representation of key elements.
2. Information from the manufacturer
- Labels and packaging – in required languages.
- Instructions for use – available in countries of sale.
3. Design and production information
- Design process – stages, specifications and validation.
- Production – data on processes, subcontractors and locations.
4. Safety and performance requirements
- Regulatory compliance – analysis, validation and documentation.
- Confirmatory methods – harmonized standards, specifications, verification of conformity.
5. Risk-benefit analysis
- Risk assessment – solutions to minimize threats.
- Benefits – analysis compliant with regulatory requirements.
6. Product verification and validation
- Tests and research – preclinical and clinical results.
- Biocompatibility data – safety analysis, compliance with standards.
- Software validation – design, development and testing.
- Stability and durability – studies on service life.
7. Additional information (in special cases)
- Active substances – if the product contains a medicinal product.
- Human/animal fabrics and cells – their origin and compliance with regulations.
- Chemicals and interactions – toxicity, absorption, metabolism.
- Sterilization and measurements – description of sterilization methods and validation of measurement accuracy.
- Compatibility with other products – evidence of compatibility in configuration with other devices.
Other issues are listed in Annexes I and II to this Regulation.
Article 61 – Clinical evaluation
- Purpose of clinical evaluation Clinical evaluation confirms the compliance of the device with the safety and effectiveness requirements, based on clinical evidence adapted to its intended use.
- Consultations with experts The manufacturer of class III and some class IIb devices may consult the clinical development strategy with a panel of experts, whose opinions should be included in the documentation.
- Scope of clinical evaluation Includes critical analysis of:
- available scientific literature if the product is equivalent to one already assessed,
- clinical trial results in accordance with regulations,
- alternative treatments.
- Requirement for clinical trials Clinical trials are mandatory for implantable and class III devices unless:
- they constitute a modification of a product already placed on the market,
- their equivalence with another product has been demonstrated, which is confirmed by a notified body.
- Product equivalence The manufacturer can claim equivalence with another product if it has full access to its technical documentation.
- Clinical Trial Exceptions Not required for devices:
- introduced in accordance with previous directives, if their clinical evaluation is in line with current requirements,
- such as sutures, staples, dental implants, etc., if based on sufficient clinical data.
- Justification for exceptions Any exception must be justified in the clinical documentation and approved by the notified body.
- Updating the list of products The European Commission may modify the list of products exempt from clinical trials.
- Clinical evaluation of non-medical products For products without a medical purpose, the clinical evaluation is based on safety and efficacy data.
- Lack of clinical data If a device does not require clinical data, the manufacturer must provide a justification based on risk management and the specificity of interaction with the human body.
- Update of the clinical evaluation Must be regularly updated based on post-market surveillance data. For class III devices and implants, the update shall take place at least once a year.
- Clinical evaluation documentation It is part of the technical documentation and for standard devices requires a clinical evaluation report.
- Uniform application The Commission may issue implementing acts to clarify divergences of interpretation.
Informed consent
Informed consent must be in writing, dated and signed by the research participant or their legal representative and the interviewer. In the case of people unable to write, it may be expressed by alternative methods in the presence of a witness.
The participant or his representative must be provided with full, clear and understandable information regarding:
- the purpose, benefits and risks of the study,
- participant rights, including the possibility of withdrawal,
- test conditions and alternative treatment methods,
- compensation system and clinical trial identification.
Information is provided in writing and is tailored to the needs of the participant. Before consent is given, it is checked whether the participant has understood the information. The participant is informed about the publication of the results of the clinical trial. In the case of minors, if they are capable of expressing an opinion, the consent of the child is required in addition to the consent of the legal representative.
Clinical trials involving individuals unable to give consent.
The study may be conducted if:
- the consent of the legal representative was obtained,
- the participant received information adapted to his/her capabilities,
- the researcher respects the participant’s will to participate in the study,
- there are no financial incentives other than reimbursement of costs,
- the study concerns the participant’s disease and the data cannot be obtained in any other way,
- there is a scientific basis to believe that the benefits outweigh the risks.
The participant should participate in the consent process to the greatest extent possible.
Clinical trials involving minors
The trial is permissible if:
- the consent of the legal representative was obtained,
- the minor received information appropriate to his or her age,
- the researcher respects the child’s will to participate,
- there are no financial incentives other than reimbursement of costs,
- the study concerns the child’s illness and is necessary for data validation,
- the benefits of participating outweigh the risks,
- the minor actively participates in the consent process,
- Once a child reaches the age to give consent, they must reconfirm their consent.
Clinical trials with pregnant and breastfeeding women
This examination is permissible when:
- the benefits outweigh the risks to the woman, fetus or child,
- health risks to the breastfed baby are avoided,
- There are no financial incentives other than reimbursement of costs.
Conformity assessment procedures
Before placing devices on the market or putting them into service, manufacturers shall carry out a conformity assessment in accordance with the relevant procedures set out in Annexes IX to XI. For class III devices, manufacturers shall apply the conformity assessment procedure in Annex IX. They may also use the procedure in Annex X in combination with the conformity assessment in Annex XI. For class IIb devices , the conformity assessment in accordance with Annex IX shall apply, covering the technical documentation of at least one representative device in each group. For class IIb implants , the assessment shall cover each device, with exceptions (e.g. sutures, staples, dental crowns). Alternatively, the type examination in Annex X and the verification of the product’s conformity in Annex XI may be used. If the technologies used are comparable to those already recognised or when the protection of public health so requires, the Commission may amend the list of class IIb implants covered by the conformity assessment. For class IIa devices, the conformity assessment shall cover the technical documentation of at least one product in the category concerned (Annex IX). Alternatively, the documentation in accordance with Annexes II and III may be drawn up and the conformity assessment in Annex XI may be used. Manufacturers of class I devices submit an EU declaration of conformity after preparing the technical documentation. If the device is sterile, has a measuring function or is a reusable surgical instrument, additional procedures apply (Annexes IX or XI), but the involvement of the notified body is limited to specific aspects, e.g. sterility or metrological requirements. Custom-made devices are subject to the procedure in Annex XIII. Class III implants additionally undergo a conformity assessment in Annex IX or XI. For specific categories of medical devices, additional procedures in Annexes IX-X also apply, e.g. for substances absorbed by the body. Conformity assessment documentation may be required in a language determined by the Member State in which the notified body operates. Investigational medical devices are subject to the provisions in Articles 62 to 81. The Commission may introduce additional rules on the application of the conformity assessment procedures, e.g. specifying the frequency of audits and random tests or minimum requirements for the assessment of the technical documentation.